洁净室及相关受控环境的专业解读

在2010年2月12日卫生部发布新版GMP并宣布3 月1 日起施行之前,我国医药行业及其主管部门,也许并不在意由中华人民共和国质量监督检验检疫总局和中国国家标准化管理委员会于同年1 月14 日联合发布的,一个同样与医药行业密切相关的国家标准,它就是“GB/T25915(2010)洁净室及相关受控环境”,一套 10 件的系列国家标准。
国标“GB/T25915(2010)洁净室及相关受控环境”使用翻译法,等同采用国际标准“ISO14644洁净室及相关受控环境”;它的第 4 部分 G B /T2 591 5. 4(201 0)-设计、建造、启动等同于 ISO14644.4(2004),是一份适用于我国航空航天、微电子、制药、医疗器械、医疗卫生等行业产品和工艺中污染物控制的国家标准。我国新版 GMP(2010)等效采用欧盟 GMP指南(2009);国标 GB/T25915(2010)等同采用国际标准 ISO14644(2004)。
新版GMP 和GB/T25915 这两份国家法规和国家标准,,都准确传递了当今国际制药行业对药品生产环境污染控制的要求。以无菌药品生产环境空气洁净度为例,欧盟(EU)、世界卫生组织(WHO)采用 ABCD模式,美国(FDA)和国际标准化组织(ISO延续 FS209 标准采用 ACD 模式(表 1)延续 FS209 标准采用 ACD 模式(表 1)
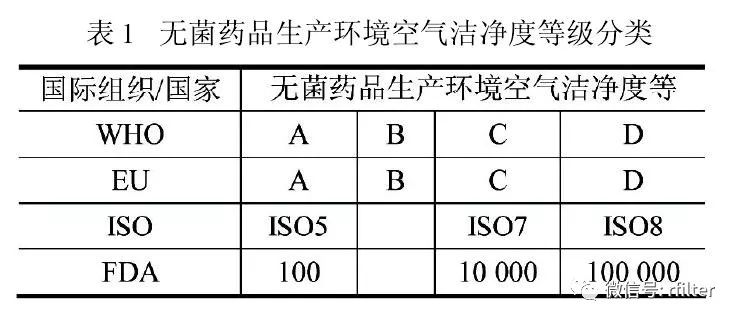
关于这两种看似不同的模式,欧盟 GMP 指南(2009)引言中有一段耐人寻味的表述:“本指南无意成为任何新概念或新技术发展的障碍,如果通过验证并证明所用方法能达到至少与本指南所述方法等价的质量保证水平,也应予以认同(TheGuide is not intended to place any restraint upon thedevelopment of any new concepts or new technologieswhich have been validated and which provide a level of Quality Assurance at least equivalent to those set out in this Guide)”。
同时,它还明确表达“药品多年来按 GMP 要求生产,没有执行 ISO 的标准。企业可自行决定采用ISO标准作为实施制药领域质量体系的一种手段(The manufacture of medicinal products has for many years taken place in accordance withguidelines for GMP and the manufacture of medicinalproducts is not implement by ISO standards. Harmo-nized standards as adopted by the ISO may be used atindustry’s discretion as a tool for implementing a quality system in the pharmaceutical sector)”。
同样,FDA 的指南文件也有类似内容的表述,足见这两种模式在国际制药行业的互认和共容。欧盟GMP指南与ISO14644.4 在控制方法上的差异,并不说明它们在控制水平上的差距。国际制药企业在实施时可自行选用,无须厚此薄彼,只要认真执行都能达到同样的质量保证水平。因此,美国制药企业产品无需按欧盟的ABCD模式方可进入欧盟国家;同样欧盟制药企业产品也没有必要改成FDA 的ACD 模式才能进入美国市场。
可是,当这两项国际标准演变成我国的法规和标准时,我国的制药企业就没有这么幸运。因为我国新版GMP在等效采用欧盟GMP指南时,删除了欧盟GMP指南的这段“提示”。新版 GMP 发布前,我国以往 GMP中无菌药品生产环境的空气洁净度标准基本采用ACD 模式。客观上,我国历版 GMP 内容和企业执行力度都与国际要求存在一定差距,追其原因错综复杂,我国无论在认知理念、综合国力、技术水平等方面都无法与国外发达国家相比,即使照搬照套国外 GMP,也不能起到立竿见影的效果。如果新版 GMP 能根据国情吸纳欧盟 GMP 指南的宽容,大多数企业可在原有基础上进行填平补齐的改造,而无需如今伤筋动骨式的重建。
这无论对国家还是对企业,都是值得深思的大事。
新版 GMP 是强制执行的国家行政法规,是制药企业认证检查的唯一标准,得不到认证证书,企业必须关停并转;而B/T25915 是国家推荐标准,又无相应检查颁证手段,自然无法与新版GMP相提并论。出现如此问题,并不说明这两项标准的水平差距,而是国家行政制度、权力上的失衡,需要由政府有关部门采取措施予以弥合修补。GB/T 25915.4(2010)-设计、建造、启动,即 ISO14644.4 的附录 B,提出了无菌产品在颗粒物和微生物受控的洁净区内无菌灌装线上灌装的空气洁净度等级要求(表 2 )。
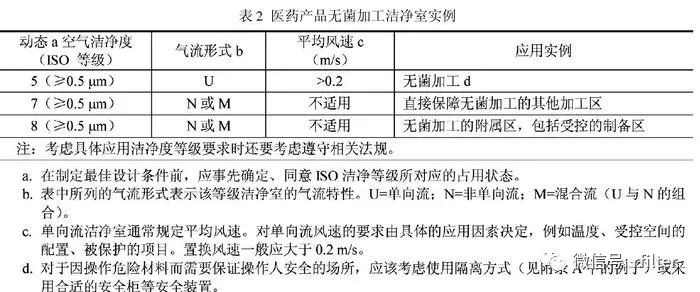
表2中值得我们注意的 ,一是空气洁净度等级的占有状态应在设计前由业主和设计人员事先商议确定。这是 ISO14644 系列标准在定义“动态”、“静态”时的一贯主张,因为各种占用状态都有它的使用价值,不能简单地以使用何种“占用状态”来评判标准的孰高孰低。受无菌药品灌装线上人员操作、设备性能、生产运行等因素影响,单向流罩下的空气洁净度往往难以测准,为确保测试状态的稳定性和判断结果的可比性,日本把测试要求原则上定为“静态”,也有国家规定动态检测范围不包括操作点或发尘点60 cm以内的区域。
我们不能单凭这些规定就此认定这些国家GMP 要求比欧盟GMP 低;二是强调单向流风速要由具体应用因素决定,给出的范围是大于 0.2 m/s,因为受保护对象要求的不同、操作状态(人员、设备、工艺、温度等)的差异,风速可在大于 0.2 m/s 的前提下调节,而不是一律“0.36 m/s~0.54 m/s”。
根据 GB/T 25915.3 单向流罩风速测点一般定在过滤器出风面150 mm~300mm 处。新版 GMP 要求的单向流风速是指工作区,它与出风口的距离至少为2 m~2.5 m,通常工作区风速为单向流罩出口风速的 60%~65%,因此出风口风速将远远大于0.36m/s- 0.54m/s才能满足工作区需要;三是为确保操作人员不受特殊药品危险性的影响,也为彻底排除操作人员对无菌灌装环境的干扰,建议采用隔离方式,如“干预受限(限制进入)的屏障系统(Restrictive Access BarrierS y s t e m ,简称 R A B S )”GB/T25915.4(2010)有着与新版 GMP 同样的控制理念,都为了有效控制药品生产环境的污染。
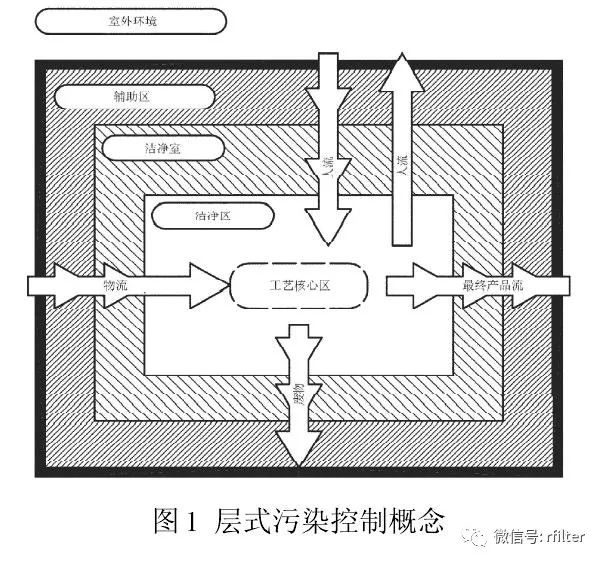
图 1 是 GB/T25915.2010)的层式污染控制概念。工艺核心区(与环境发生交互作用的工艺位置)所在的区域为洁净区,是洁净室中受控最严的部分,空气洁净度等级为单向流100 级(即ISO 5 级,欧盟 A 级)。出于经济、技术和运行等方面的原因,要尽量减小最高洁净度区域的范围。洁净区www.iwuchen.com通常采用密闭式(如 RABS),或由洁净度较低的外部区域-洁净室(1 万级,即 ISO7 级)包围。相邻区域间的人流、物流进入工艺核心区时,会增加传播污染的风险,因此应特别注意人流和物流的布局细节与管理,这就是层式污染控制的基本概念。

与 GB/T25915.4(2010)不同的是新版GMP将包围洁净区的外部区域(洁净室)的空气洁净度等级设定为B级,所谓B级是静态控制时相同于A 级(单向流 100 级)的静态,动态控制时相同于 C 级(1 万级)的静态。GB/T 25915.4(2010)和新版 GMP 都认为无菌灌装线外部区域(洁净室)空气洁净度等级应低于工艺核心区(洁净区),至于低多少,是1 万级还是B 级?这是可探讨的学术问题,不应成为判定标准高低的分界线。
国内外实践证明,背景设置为1 万级或B 级,都能保护无菌药品的生产环境,我们应从分析影响无菌灌装线的环境因素着手,寻找控制污染源的途径,才能最终确保无菌生产环境。
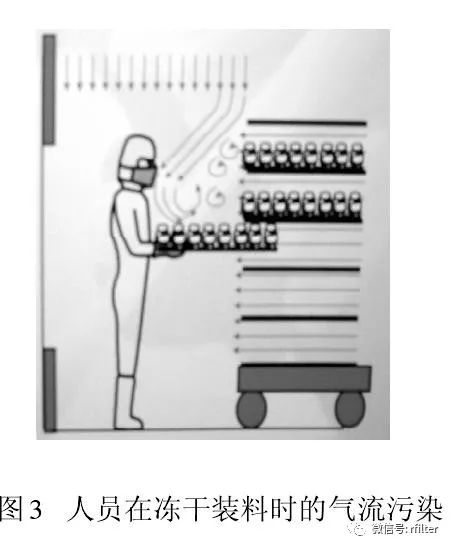
从人们开始接受并应用洁净技术时就发现,人是洁净室最大的污染源。以微粒和微生物为污染物控制对象的制药生产,大量测试数据和生产实践告诫我们,人员(生产、检验、管理、维修)因素无时无刻不在干扰药品生产。图2 显示人员在单向流下操作导致气流粒子污染。此刻,保护性的单向流变成污染物的载体,直接影响被保护的产品。此时气流速度越大,则污染程度越大。图3 为冻干剂装料时,单向流罩下垂直和水平气流相互冲突,形成紊流,紊流气流会在较长时间里产生悬浮污染。不言而喻,人是无菌灌装过程中影响最大的环境因素。
为此,实现无菌灌装环境控制的技术要点是:
(1)将人员从无菌环境隔离,从根本上排除人的干扰;
(2)采取局部隔离无菌环境 (如R A B S 等);
(3 )即便如此仍需限制人员对无菌产品的干预;
(4)生产运行中不让人员进入无菌环境。不解决人对无菌环境干扰问题,大幅度提高无菌药品生产线外部区域空气洁净度等级,效果也不会明显。
必须强调,医药工程设计中空气净化措施只是控制洁净室微生物的手段之一,这些措施只能控制通过空气途径传播的微生物污染,对表面对象微生物的生物污染以及化学污染都无能为力,药品生产使用的设备、设施、容器具、物料、包装材料等沾附的微生物、化学品,以及生产、管理人员携带的微生物,只有采用清场、清洗、消毒、灭菌等措施,任何级别的净化空调都无法替代。
近年来国外对空气净化的滤菌效果提出质疑。数据表明 1 台 1 m2高效过滤器,在 0.45 m/s 流速下 1 h 可能漏过去的细菌约 1620 cfu,8 h 就高达12960 cfu。在适合微生物生长的环境下,它们还会以几何级数疯长。仅采用高效过滤器营造无菌环境真的安全可靠吗?空气洁净技术的核心是过滤技术,并不包括消毒杀菌和控制化学污染。
新版GMP对动态环境空气无菌品质要求提出了严格的控制指标,,然而,单纯的传统空气洁净技术与管理难以实现动态A级要求。国外关于空气无菌净化技术的新发展,为我们实现动态A 级生产环境提供了新思路,如管道紫外杀菌技术、光催化离子杀菌技术、低温等离子技术以及种种物理和化学复合杀菌技术等,不但可有效杀灭空气中的微生物,满足动态A级微生物控制指标,提升制药企业药品生产环境的无菌水平,而且对表面微生物和化学污染同样能取得满意的效果。如果制药企业的 GMP 改造,不在空气无菌净化技术上采取措施,继续走传统空气洁净技术老路,将会大大浪费洁净室建设费用和运行成 本 。
国标 GB/T 25915.4(2010)的内容包括规划和设计、建造和启动、检测和验收、文件等方面要求,涵盖工程建设的全过程,对我国医药行业尤为重要。药品生产实施 GMP 是一项系统工程,为药品生产提供符合 GMP 要求的厂房设施是企业实施G M P 的先决条件。
因此,医药工程项目必须按GMP 要求实施规范管理,才能为企业实施 GMP提供可靠保障。近年来国外针对医药工程项目的规范化管理,一项新的管理规范— — GEP已在一些发达国家悄然问世。GEP(医药工程质量管理规范)是Good Engineering Practice 的缩写,是与GMP配套的药品生产管理文件。它的宗旨是“将已确立的工程设计方案和标准应用于项目的整个生命周期中,提供合适成本的解决方案”。我国至今尚无这方面的规范,工程项目大多由企业凭经验自行管理,主要依据是各类相关的国家标准,缺乏系统性、协调性。
国标 GB/T 25915.4(2010)的发布,为工程项目规划、设计、施工、试车、检测、验收等环节的规范化管理填补了空白,国标所附的大量资料性附录,如“设施的验收”、“设施的布局”、“建造和材料”、“洁净室的环境控制”、“待需方/ 用户与供方/ 设计方商定的补充技术要求”等,为项目的工程设计、采购供应、施工安装、试车验收、工程进度、技术质量、安全环保、资金运作、部门协调等提供了详尽的规范要求,制药企业应自觉以国标GB/T 25915为准绳,并据此制订医药工程项目的GEP,确保工程项目符合 GMP 要求。
国标GB/T 25915系列标准反映了当今国际“洁净室及相关受控环境 ”的新概念、新思路和新方法,是指导我国各行各业洁净技术的规范性文件,必将成为我国洁净技术蓬勃发展的新起点。
【来源】互联网
【声明】部分文章和数据信息来源于互联网。如转载内容涉及版权等问题,请立即与我们联系,我们将迅速采取适当措施。