NMPA发布:医疗器械重点监管目录(附清单)

9月9日,国家药监局综合司发布了《关于加强医疗器械生产经营分级监管工作的指导意见》。这份文件的重要性之一在于,它将指导各地药品监管部门在医疗器械注册人制度下进行医疗器械生产经营监管。
《指导意见》要求省级药品监督管理部门和设区的市级负责药品监督管理的部门结合产业发展和监管实际,再制定符合本地区实际的生产和经营分级监管细化要求。
特别注意的是 ,文件提出了对医疗器械生产经营企业的监管级别划分原则和检查要求,药品监管部门可以按照风险将医疗器械企业划分为四个监管级别,对不同监管级别的企业实施相应监管措施。
即:对于长期以来监管信用状况较好的企业,可以酌情下调监管级别;对于跨区域委托生产的医疗器械注册人,仅进行受托生产的受托生产企业,以及异地增设库房的经营企业等,应当酌情上调监管级别。对医疗器械生产企业采取非预先告知的方式进行监督检查,对经营企业采取突击性监督检查。
这份《指导意见》自2023年1月1日起施行。下文中01和02为生产端,03和04位经营端。
01再次细化:各个地区将出台监管目录
本次通知中最受关注和讨论的焦点就是“分级监管”。
首先是将落实生产分级监管职责。国家药品监督管理局负责指导和检查全国医疗器械生产分级监管工作,制定医疗器械生产重点监管品种目录。
省、自治区、直辖市药品监督管理部门负责制定本行政区域医疗器械生产重点监管品种目录,并组织实施医疗器械生产分级监管工作;设区的市级负责药品监督管理的部门依法按职责负责本行政区域第一类医疗器械生产分级监管的具体工作。
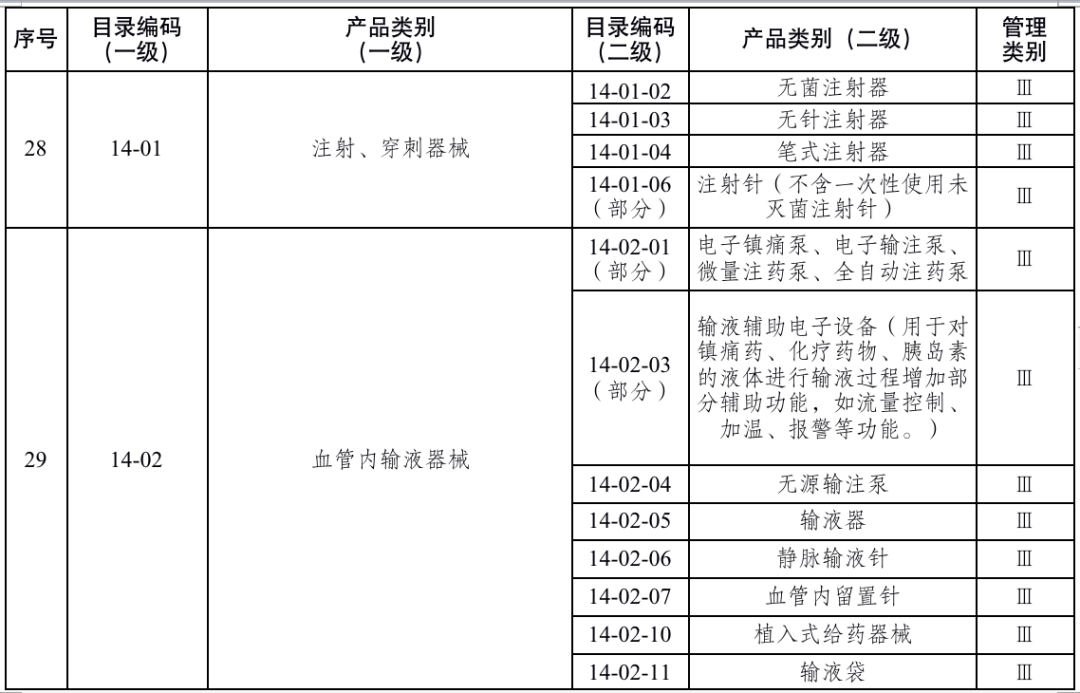
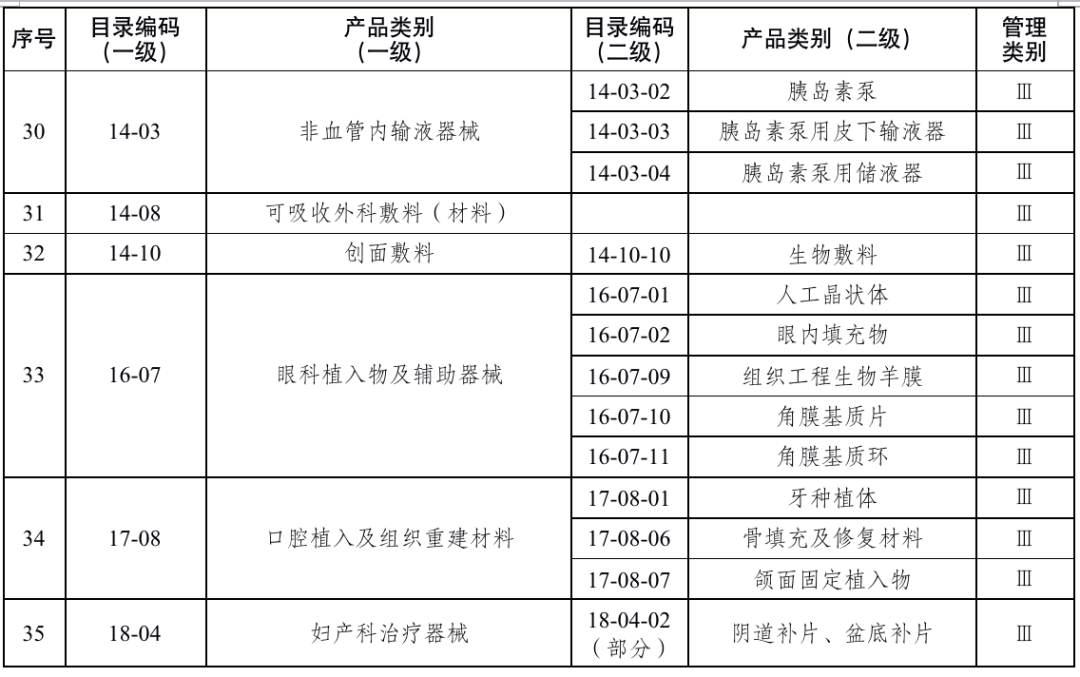
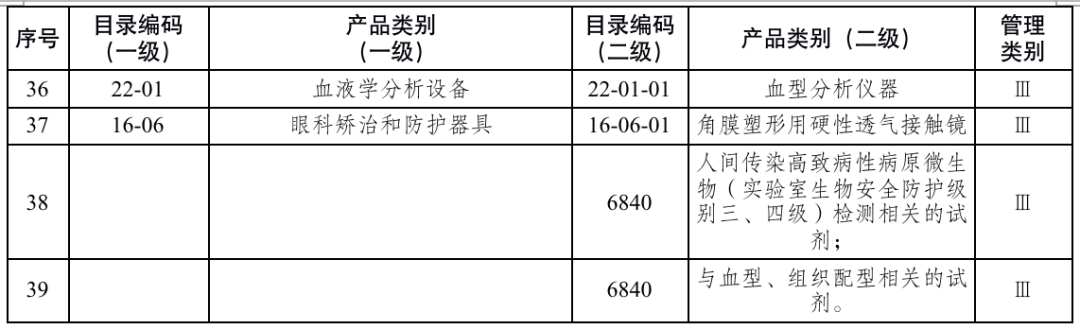
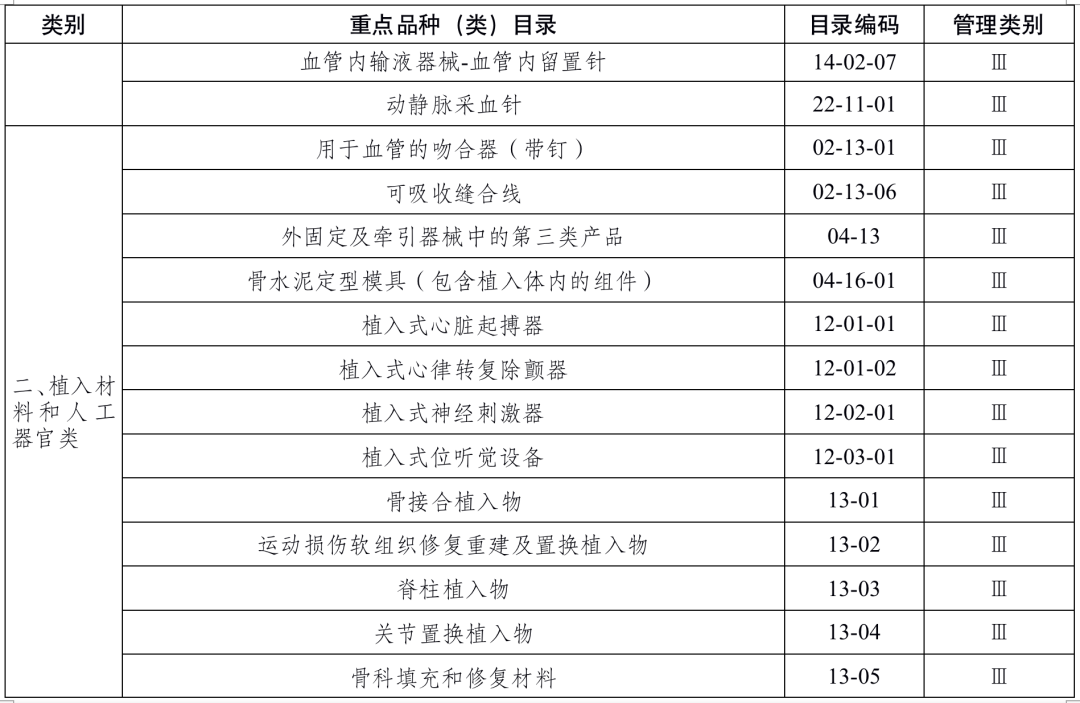
国家药品监督管理局根据医疗器械产品风险程度制定并动态调整医疗器械生产重点监管品种目录(见文末 附件1);省、自治区、直辖市药品监督管理部门应当综合分析本行政区域同类产品注册数量、市场占有率、生产质量管理总体水平和风险会商情况等因素,对国家药品监督管理局制定的目录进行补充,确定本行政区域医疗器械生产重点监管品种目录并进行动态调整。
对于跨区域委托生产的医疗器械注册人,由注册人所在地省、自治区、直辖市药品监督管理部门负责研究确定其产品是否纳入本行政区域医疗器械生产重点监管品种目录。
省、自治区、直辖市药品监督管理部门应当结合本行政区域产业发展、企业质量管理状况和监管资源配备情况,制定并印发医疗器械生产分级监管细化规定,明确监管级别划分原则,以及对不同监管级别医疗器械注册人备案人、受托生产企业的监督检查形式、频次和覆盖率。
那么,监管级别划分和检查要求会按照何种原则执行?
对风险程度高的企业实施四级监管,主要包括生产本行政区域重点监管品种目录产品,以及质量管理体系运行状况差、有严重不良监管信用记录的企业;对风险程度较高的企业实施三级监管,主要包括生产除本行政区域重点监管品种目录以外第三类医疗器械,以及质量管理体系运行状况较差、有不良监管信用记录的企业;对风险程度一般的企业实施二级监管,主要包括生产除本行政区域重点监管品种目录以外第二类医疗器械的企业;对风险程度较低的企业实施一级监管,主要包括生产第一类医疗器械的企业。
这里需要注意的是,涉及多个监管级别的,按照最高级别进行监管。
一般情况下,对实施四级监管的企业,每年全项目检查不少于一次;对实施三级监管的,每年检查不少于一次,其中每两年全项目检查不少于一次;对实施二级监管的,原则上每两年检查不少于一次;
对实施一级监管的,原则上每年随机抽取本行政区域25%以上的企业进行监督检查,并对新增第一类医疗器械生产企业在生产备案之日起3个月内开展现场检查,必要时对生产地址变更或者生产范围增加的第一类医疗器械生产企业进行现场核查。
监督检查可以与产品注册体系核查、生产许可变更或者延续现场核查等相结合,提高监管效能。
全项目检查是指药品监督管理部门按照医疗器械生产质量管理规范及相应附录,对监管对象开展的覆盖全部适用项目的检查。对委托生产的医疗器械注册人备案人开展的全项目检查,应当包括对受托生产企业相应生产活动的检查。
02动态调整级别:增加哪些关键监管维度?
在动态调整监管级别方面。国家药监局指出:省、自治区、直辖市药品监督管理部门应当根据医疗器械生产分级监管细化规定,结合监督检查、监督抽验、不良事件监测、产品召回、投诉举报和案件查办等情况,每年组织对本行政区域医疗器械注册人备案人、受托生产企业风险程度进行科学研判,确定监管级别并告知企业。
对于当年内医疗器械注册人备案人、受托生产企业出现严重质量事故,新增高风险产品、国家集中带量采购中选产品、创新产品等情况,应当即时评估并调整其监管级别。
对于长期以来监管信用状况较好的企业,可以酌情下调监管级别;
对于以委托生产方式或者通过创新医疗器械审评审批通道取得产品上市许可,以及跨区域委托生产的医疗器械注册人,仅进行受托生产的受托生产企业,国家集中带量采购中选产品的医疗器械注册人备案人、受托生产企业应当酌情上调监管级别。
具体调整方式由省、自治区、直辖市药品监管部门结合本行政区域企业整体监管信用状况、企业数量和监管资源配比等情况确定。
国家药监局强调:将根据监管级别强化监督检查。
省、自治区、直辖市药品监督管理部门应当按照分级监管规定,制定年度监督检查计划,明确检查频次和覆盖率,确定监管重点;坚持问题导向,综合运用监督检查、重点检查、跟踪检查、有因检查和专项检查等多种形式强化监督管理。监督检查可以采取非预先告知的方式进行,重点检查、有因检查和专项检查原则上采取非预先告知的方式进行。
对于通过国家药品监督管理局创新医疗器械审评审批通道取得产品上市许可的医疗器械注册人及其受托生产企业,应当充分考虑创新医疗器械监管会商确定的监管风险点和监管措施;
对于因停产导致质量管理体系无法持续有效运行的企业,应当跟踪掌握相关情况,采取有针对性的监管措施。
03经营层面:跨区域监管有哪些新要求?
国家药监局强调,将落实经营分级监管职责。
国家药品监督管理局负责指导和检查全国医疗器械经营分级监管工作,并制定医疗器械经营重点监管品种目录;省、自治区、直辖市药品监督管理部门负责指导和检查设区的市级负责药品监督管理的部门实施医疗器械经营分级监管工作;设区的市级负责药品监督管理的部门负责制定本行政区域医疗器械经营重点监管品种目录,组织实施医疗器械经营分级监管工作;县级负责药品监督管理的部门负责本行政区域内医疗器械经营分级监管具体工作。
对于跨设区的市增设库房的医疗器械经营企业,按照属地管理原则,由经营企业和仓库所在地设区的市级负责药品监督管理的部门分别负责确定其监管级别并实施监管工作。
结合实际确定重点监管品种目录。
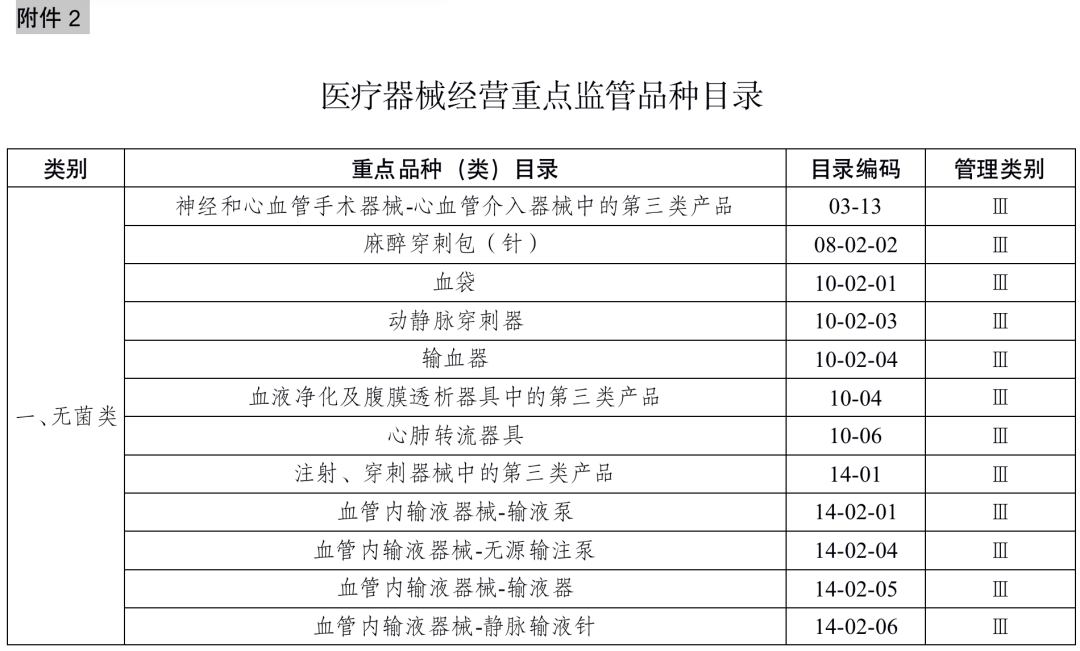
国家药品监督管理局根据医疗器械产品和产品经营风险程度,制定并动态调整医疗器械经营重点监管品种目录(见文末附件2);设区的市级负责药品监督管理的部门应当综合分析产品监督抽验、不良事件监测、产品召回、质量投诉、风险会商情况等因素,对国家药品监督管理局制定的目录进行补充,确定本行政区域医疗器械经营重点监管品种目录并进行动态调整。
对于跨设区的市增设库房的医疗器械经营企业,由库房所在地设区的市级负责药品监督管理的部门负责确定其库存的产品是否属于本行政区域医疗器械经营重点监管产品。
设区的市级负责药品监督管理的部门应当根据本行政区域医疗器械经营的风险程度、经营业态、质量管理水平和企业监管信用情况,结合医疗器械不良事件及产品投诉状况等因素,制定并印发分级监管细化规定,明确监管级别划分原则,以及对不同监管级别医疗器械经营企业的监督检查形式、频次和覆盖率。
监管级别划分和检查要求按照以下原则进行:
对风险程度高的企业实施四级监管,主要包括“为其他医疗器械注册人、备案人和生产经营企业专门提供贮存、运输服务的”经营企业和风险会商确定的重点检查企业;
对风险程度较高的企业实施三级监管,主要包括本行政区域医疗器械经营重点监管品种目录产品涉及的批发企业,上年度存在行政处罚或者存在不良监管信用记录的经营企业;
对风险程度一般的企业实施二级监管,主要包括除三级、四级监管以外的经营第二、三类医疗器械的批发企业,本行政区域医疗器械经营重点监管品种目录产品涉及的零售企业;
对风险程度较低的企业实施一级监管,主要包括除二、三、四级监管以外的其他医疗器械经营企业。
涉及多个监管级别的,按最高级别对其进行监管。
实施四级监管的企业,设区的市级负责药品监督管理的部门每年组织全项目检查不少于一次;实施三级监管的企业,设区的市级负责药品监督管理的部门每年组织检查不少于一次,其中每两年全项目检查不少于一次;实施二级监管的企业,县级负责药品监督管理的部门每两年组织检查不少于一次,对角膜接触镜类和防护类产品零售企业可以根据监管需要确定检查频次;
实施一级监管的企业,县级负责药品监督管理的部门按照有关要求,每年随机抽取本行政区域25%以上的企业进行监督检查,4年内达到全覆盖。必要时,对新增经营业态的企业进行现场核查。
全项目检查是指药品监督管理部门按照医疗器械经营质量管理规范及相应附录,对经营企业开展的覆盖全部适用项目的检查。对“为其他医疗器械注册人、备案人和生产经营企业专门提供贮存、运输服务的”经营企业开展的全项目检查,应当包括对委托的经营企业的抽查。
动态调整监管级别。设区的市级负责药品监督管理的部门应当根据医疗器械经营分级监管细化规定,在全面有效归集医疗器械产品、企业和监管等信息的基础上,每年组织对本行政区域医疗器械经营企业、跨设区的市增设库房的医疗器械经营企业进行评估,科学研判企业风险程度,确定监管级别并告知企业。对于新增经营业态等特殊情况可以即时确定或调整企业监管级别。
对于长期以来监管信用情况较好的企业,可以酌情下调监管级别;对于存在严重违法违规行为、异地增设库房、国家集中带量采购中选产品和疫情防控用产品经营企业应当酌情上调监管级别。具体调整方式由设区的市级负责药品监管的部门结合本行政区域企业整体监管信用状况、企业数量和监管资源配比等情况确定。
根据监管级别强化监督检查。地方各级负责药品监督管理的部门应当根据监管级别,制定年度监督检查计划,明确检查重点、检查方式、检查频次和覆盖率。检查方式原则上应当采取突击性监督检查,鼓励采用现代信息技术手段实施监督管理,提高监管效率和水平。
备注:本指导意见施行后。原国家食品药品监督管理总局《关于印发〈医疗器械生产企业分类分级监督管理规定〉的通知》(食药监械监〔2014〕234号)、《关于印发国家重点监管医疗器械目录的通知》(食药监械监〔2014〕235号)、《关于印发〈医疗器械经营企业分类分级监督管理规定〉的通知》(食药监械监〔2015〕158号)和《医疗器械经营环节重点监管目录及现场检查重点内容》(食药监械监〔2015〕159号)同时废止。
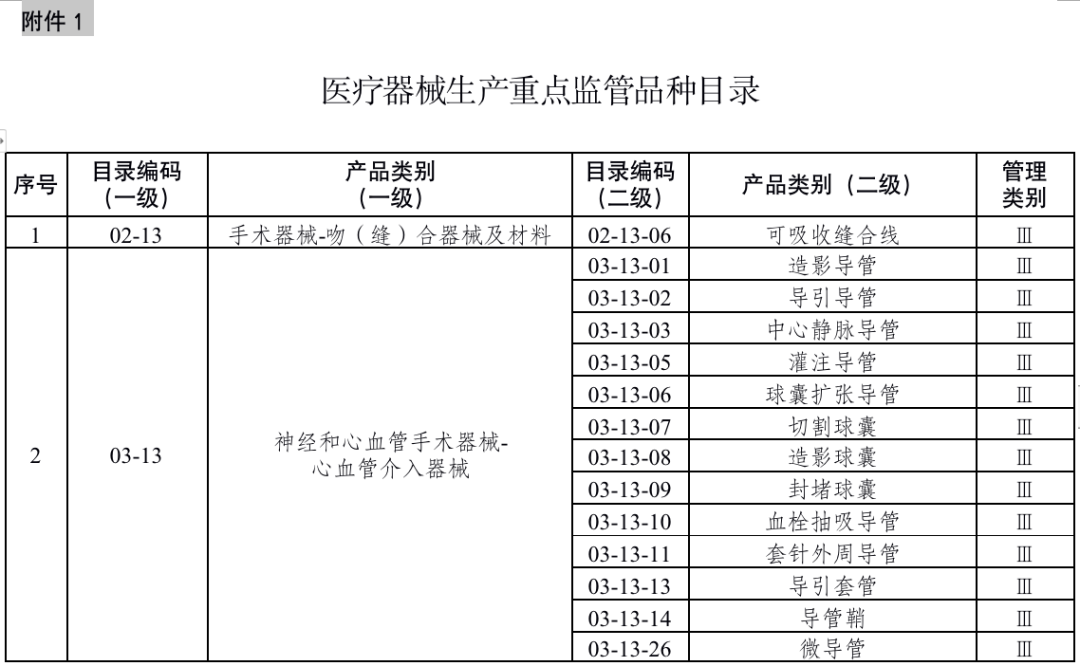
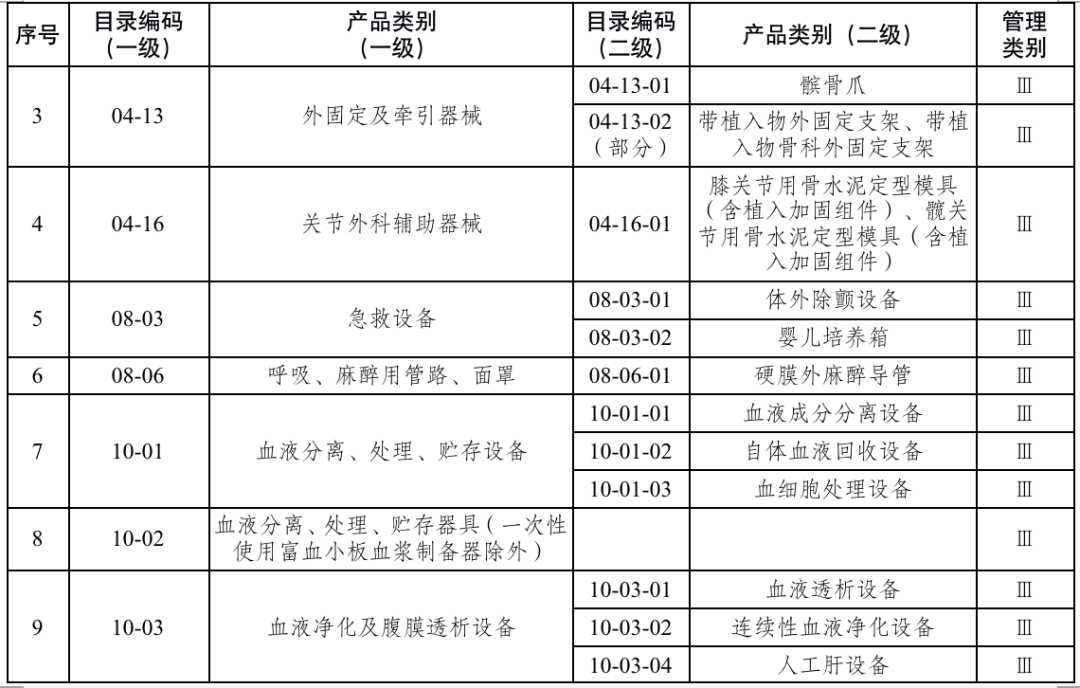
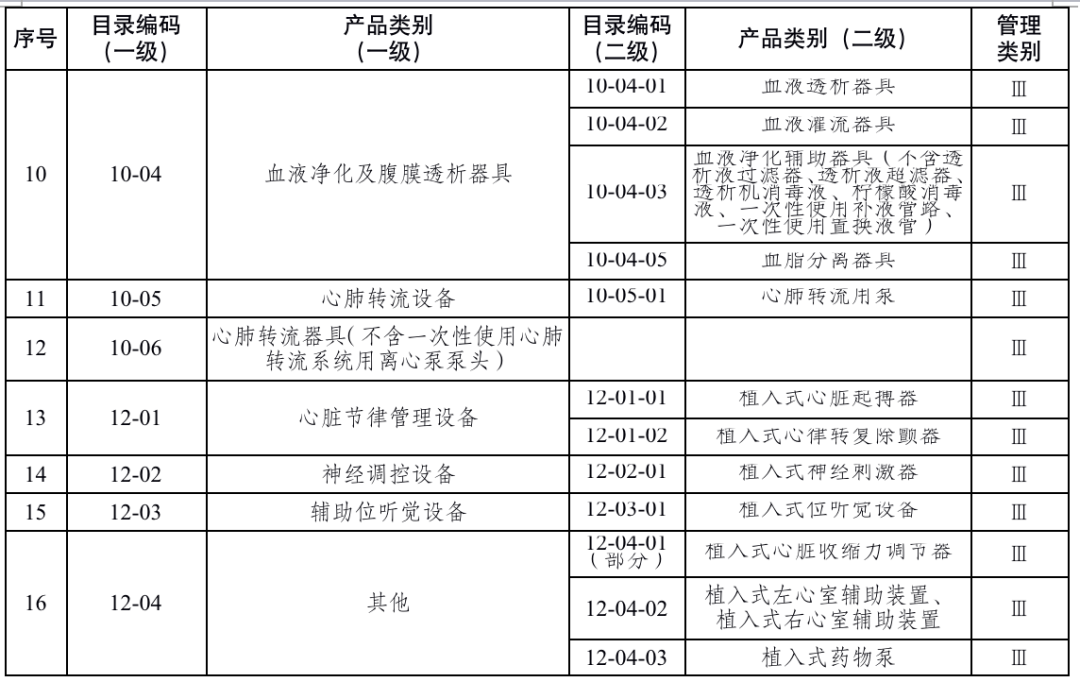
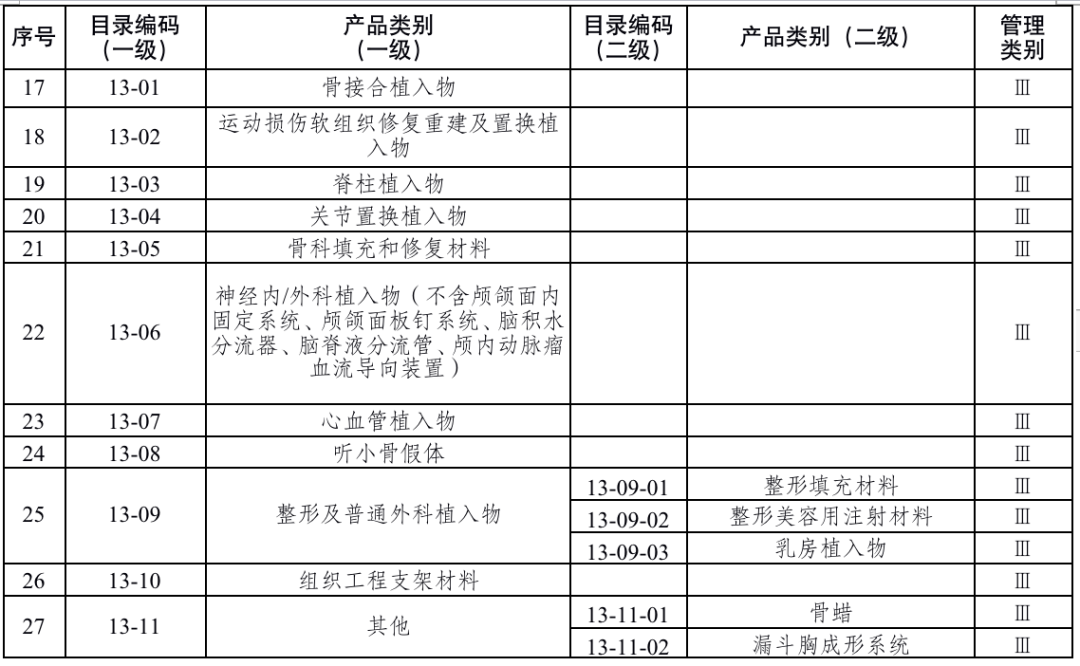


来源:国家药监局
【文章内容与信息来源于互联网或转载,我公司不对本文所包含内容的准确性、可靠性或者完整性提供任何明示或暗示的保证,不对本文观点负责。如转载内容涉及版权等问题,请立即与我们联系,我们将迅速采取适当措施,以保障双方权益,谢谢。】
注:文章配图为网络转载图片,侵权即删!